
Research on a key brain immune cell suggests it is a tantalizing but slippery target for new therapies
Peer inside the brain of someone with Alzheimer’s disease, and you’ll see some striking features: shriveled nerve cells and strange protein clumps. According to a leading theory, proteins called amyloid beta and tau build up in the brain and choke nerve cell communication, setting the disease in motion years before people suspect anything is wrong with their recall.
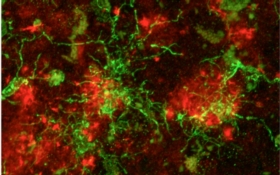
Yet the Alzheimer’s brain has another curious aspect. Some of the clusters of toxic amyloid proteins are entangled with octopus-like immune cells called microglia, cells that live in the brain to clear unwanted clutter. By munching on amyloid plaques, microglia are thought to help keep the disease at bay. But these housekeeping cells have an additional role—they switch on inflammatory pathways. Inflammation is critically important when the immune system encounters infection or needs to repair tissue. If left unchecked, however, the inflammatory process churns out toxic substances that can kill surrounding cells, whose death triggers more inflammation and creates a vicious cycle.
For years scientists have probed how neuroinflammation contributes to Alzheimer’s disease and other neurodegenerative ailments. Researchers face a number of immediate questions: Is neuroinflammation a driving force? Does it kick in when the disease is already underway and worsen the process? Could it be harnessed for good in the early stages? Those questions are far from settled, but research is starting to reveal a clearer picture. “It may not be the amyloid plaques themselves that directly damage neurons and the connections between them. Rather, it may be the immune reaction to the plaques that does the damage,” says Cynthia Lemere, a neuroscientist at Brigham and Women’s Hospital. Still, it is hard to say if microglia are good guys or bad, making it challenging to create therapeutics that target these cells.
For decades multiple lines of evidence have given scientists a hunch that inflammation plays some role in Alzheimer’s. In several epidemiological studies people who regularly took ibuprofen, aspirin or other nonsteroidal anti-inflammatory drugs (NSAIDs) had lower Alzheimer’s risk than those who did not—leading some researchers to wonder if inflammation could contribute to the disease. But in a more rigorous study—which randomly assigned participants to take either NSAIDs or a placebo daily for several years and administered annual cognitive tests—anti-inflammatories did not seem to help.
Some researchers continue to believe inflammation plays a part, however. Lab experiments have shown that NSAIDs reduce brain amyloid in mice engineered to mimic some disease features. Another analysis directly linked the usage of anti-inflammatory drugs with calmer microglia as judged under a microscope. And in recent years the idea of an inflammation–Alzheimer’s link has gotten a boost from large-scale genetic analyses. Using an approach called a genome-wide association study, researchers scanned complete sets of DNA in thousands of participants, looking for small variations that appear more often in people with the disease than in healthy individuals. Such analyses have tied Alzheimer’s risk to several genes involved in innate immunity—the body’s immediate defenses that indiscriminately attack pathogens and other foreign molecules and cells. One key gene encodes the making of a receptor called TREM2, a docking site for molecules on the surface of microglia and other innate immune cells. TREM2 piqued the interest of scientists because one relatively rare version triples a person’s risk of Alzheimer’s. The past few years have seen a flurry of studies aimed at figuring out what TREM2 does. “There’s been almost nothing clear-cut about it,” Lemere says. “People were very confused.”
TREM2 initially appeared to have a protective role. In a culture dish microglia that were modified to make a lot of TREM2 gobbled more amyloid and removed more dying neurons, compared with microglia having less of the protein. And in a mouse model of Alzheimer’s, having more TREM2 in the brain seemed to relieve inflammation and amyloid buildup, slow the loss of neurons and preserve the animals’ cognitive abilities. Inconsistencies arose, however, when scientists studied mice that mimic features of Alzheimer’s and were then further genetically modified to lack TREM2. Experiments with these mice suggest TREM2 is important for steering microglia toward amyloid and turning on genes that rev up their cleanup capabilities. Curiously, though, one strain racked up tons of brain amyloid whereas another ended up with less than usual.
When researchers looked at TREM2 in people, the story again turned murky. (TREM2 straddles a cell’s membrane but enzymes snip off the protein’s external portion and release it into the cerebrospinal fluid, where researchers can measure it.) Comparing fluid samples from healthy elderly subjects and those with Alzheimer’s, one team of scientists found less TREM2 in the Alzheimer’s group whereas other groups reported its levels were higher. Last month one of those groups published a new analysis that sheds light on the confusion. As it turns out, TREM2 levels vary depending on how far the disease has progressed. In the recent analysis TREM2 levels peaked in early-stage Alzheimer’s, when people have mild memory loss, but by the time they develop full-blown dementia, TREM2 has dropped to normal levels. Using brain imaging, another team reported results in March that appear consistent with the newest TREM2 analysis. These researchers deployed positron emission tomography (PET), which measures cellular metabolism, to scan the brains of 32 healthy older adults and 64 with Alzheimer’s at various stages. The PET scans measured TSPO, a protein researchers consider a marker for neuroinflammation, because it is found at high levels on active microglia. To gauge how far the disease had progressed in participants, researchers also measured brain amyloid levels and gave cognitive tests. The findings indicate that microglia are most active in the early stages of Alzheimer’s and play a protective role during this period.
Researchers have come up with several ideas about how these cells’ activities relate to Alzheimer’s. One theory suggests microglia are helpful early in disease by gobbling amyloid but slow down as people get older. Eventually they become overwhelmed with the task and reach a point where “all they do is send out alarm signals,” says Tony Wyss-Coray, a neuroscientist at Stanford University School of Medicine. Those alarm signals are what researchers call chronic inflammation, which harms tissue and exacerbates disease. In essence, as we age, “microglia are trying hard to do their job but can’t keep up,” Wyss-Coray says.
But that begs the question: Do microglia actually drive the disease or do they act as accomplices, hastening a harmful process initiated by amyloid? Several research groups have tried to clarify this issue using various methods to clear microglia from the brains of Alzheimer’s mice to see how this would affect amyloid buildup. Surprisingly, a 2009 study by researchers in Germany found that getting rid of microglia had no effect on plaques. This suggested that “microglia were not eating and clearing the amyloid from the brain—a role they are traditionally thought to play,” says neurobiologist Kim Green of the University of California, Irvine.
The 2009 study came with a caveat, though. It used a surgical procedure that not only cleared microglia but also mucked up the mice’s brains and behavior in general. Green and co-workers worked out a gentler approach using druglike molecules to kill microglia in the brains of mice with simulated Alzheimer’s disease. The results were the same. The mice had just as much amyloid in their brains regardless of whether or not microglia were around.
By other measures, though, getting rid of microglia clearly helped. Normally Alzheimer’s-like mice lose a lot of neurons and do worse on memory tests. Animals without microglia, however, did not encounter these problems—despite having heads full of amyloid.
Recent mouse experiments by Lemere and others also suggest that microglia act not only independently of amyloid, but earlier. Last month, researchers led by Beth Stevens of Boston Children’s Hospital reported that a process in which microglia prune excess synapses in the brain during early life can turn on inappropriately later on, possibly triggering Alzheimer’s or other disorders marked by damage to connections between brain cells. The researchers showed that the synapse-eating process requires a protein in the complement system—a part of the immune response that helps “tag” unwanted cells and other debris for destruction. Lemere’s team looked at the effects of another complement protein in older Alzheimer’s-like mice with advanced disease. They saw that mice lacking this protein were protected from synapse loss via similar pruning mechanisms or other pathways that protect neuron health.
Given recent findings that paint a picture of microglia being more of a culprit than a defender, researchers might conclude that once Alzheimer’s sets in, microglia should be removed or switched off. However, “we need to keep in mind that timing and context are critically important,” says Terrence Town, a neuroscientist at the University of Southern California. Even though microglia do not appear helpful—in mice they do not even gobble amyloid in early stages of disease—that does not mean they cannot help under the right conditions by morphing back into their cleanup role, Town says.
Town and others have proposed a second, newer theory for harnessing microglia and the innate immunity to fight Alzheimer’s. Research has shown that both people with Alzheimer’s and mice used to study the disease activate powerful anti-inflammatory molecules in the brain in an attempt to quiet microglia. Ironically, though, this doesn’t help. Rather, it leads to low-level chronic inflammation that prevents microglia from being able to chew up plaques. Therefore, therapeutic efforts should not aim to get rid of microglia but rather restore proper function to these cells, Town suggests. “The focus shouldn’t be on making them anti-inflammatory or pro-inflammatory. The focus should be on bringing them back to health.”
This idea finds support from “Siamese twin” experiments Wyss-Coray and co-workers have done in mice. Using a surgical technique called parabiosis to join the circulatory system of a young mouse with that of an old one, the researchers studied the effects of young blood on elderly cells, and vice versa. Several years ago researchers measured the older animals’ production of new neurons—a process that usually diminishes with age—and found it could be rejuvenated by young blood.
More recently the team took a closer look at the microglia in the older mice. “They’re completely messed up. They are full of undigested material and not functioning the way they should,” Wyss-Coray says. But when the researchers exposed the aging microglia to young blood through parabiosis, they were reactivated. “They look more like younger microglia,” he says.
Microglia may be only one piece of the story in the Alzheimer’s disease process. But their prominence at sites where nerve cells are damaged by the disease means they deserve careful scrutiny in the desperate search for ways to arrest the most salient cause of dementia. Says Green: “Microglia are front and center in the disease and potentially a therapeutic target.Source: Uncovering New Players in the Fight Against Alzheimer’s










Leave A Reply